 预存有礼
预存有礼



GC-MS(Gaschromatography-mass spectrometry)全称气相色谱-质谱联用,简称气质联用仪。被分析样品经毛细管柱分离,进入离子源。采用电子电力标准配置(EI),产生正离子,在推斥、聚焦、引出电极的作用下将正离子送入四极杆系统。四极杆在高频电压与正负电压联合作用下形成高频电场,在扫描电压作用下,只有符合四极场运动方程的离子才能通过四极杆对称中心到达离子检测器,再经离子流放大器放大,产生质谱信号。得到了质谱图,通过解释谱图或进行谱库检索以识别未知样品的组成。
GC-MS总体上由以下五大部分组成:色谱仪(常压)、接口部分、质谱分析器(高真空)和计算机数据处理系统。示意图如图下图所示:

质谱分析器基本部件有:离子源、滤质器、检测器三部分组成,它们被安放在真空总管道内。在GC-MS联用中经过气相色谱分离的各气态分子受离子源轰击,电解裂解成分子离子,并进一步碎裂为碎片离子。在电场和磁场综合作用下,按照m/z大小进行分离,到达检测器检测、记录和整理,得到质谱图,实现样品定性定量分析。
(1)进样系统:GC出来的样品直接进入MS分析仪。
(2)离子源:离子源的作用是接受样品产生离子。常用的离子化方式有:电子轰击EI;化学电离CI。
(3)质量分析器:其作用是将电离室中生成的离子按质荷比(m/z)大小分开,进行质谱检测。常见质量分析器有:四极质量分析器; 扇形质量分析器; 双聚焦质量分析器; 离子阱检测器。
(4)检测器:检测器的作用是将离子束转变成电信号,并将信号放大,常用检测器是电子倍增器。
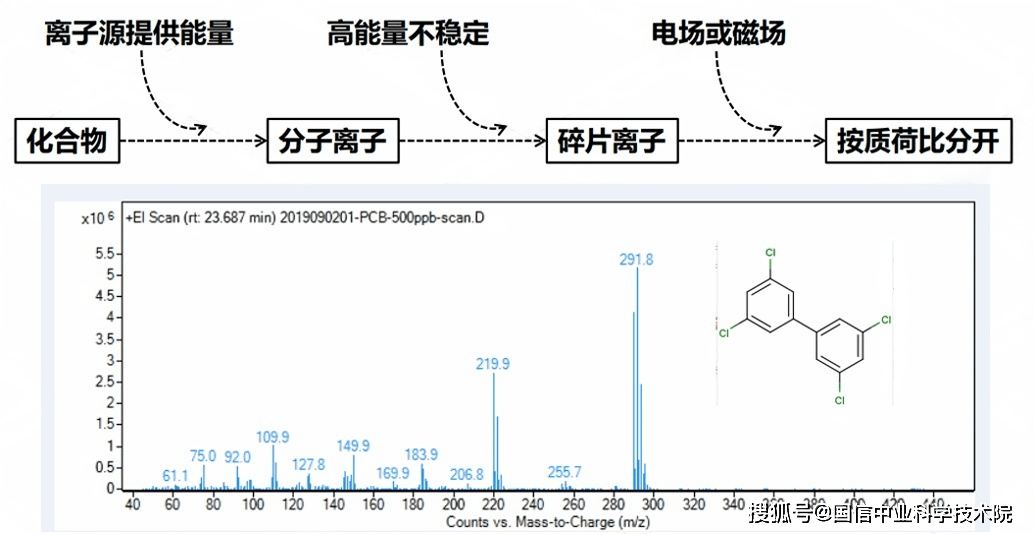
质谱(MS):按照离子的质荷比(m/z)来进行分析得到图谱的一种分析方法
GCMS的测定方法
1、总离子流色谱法(totalionizationchromatography,TIC)
类似于GC图谱,用于定量
a.反复扫描法(repetitivescanningmethod,RSM)
按一定间隔时间反复扫描,自动测量、运算,制得各个组分的质谱图,可进行定性。
b.质量色谱法(masschromatography,MC)
记录具有某质荷比的离子强度随时间变化图谱。在选定的质量范围内,任何一个质量数都有与总离子流色谱图相似的质量色谱图。
2、选择性离子监测(selectedionmonitoring,SIM)
对选定的某个或数个特征质量峰进行单离子或多离子检测,获得这些离子流强度随时间的变化曲线。其检测灵敏度较总离子流检测高2~3个数量级。
GCMS的应用范围
GCMS能检测的什么类型的化合物?
小分子:分子量小于1000 烃类:卤代烃/多环芳烃
容易气化:沸点不高,小于350℃ 有机酸:甲酸/乙酸/丙酸/丁酸等
热稳定:高温不容易发生反应
GCMS能检测哪些种类化合物?
醇:胆固醇
酚:苯酚/二氯苯酚/三氯苯酚
酯:脂肪酸甲酯/邻苯二甲酸酯
酮:直链脂肪酮/环丁酮
醛:丁醛/戊醛
醚:多溴联苯醚
含氮化合物:硝基苯/硝基苯酚
含硫磷化合物:甲硫醚类/有机磷
含卤素化合物:多氯联苯/多溴联苯
定性分析基础:
GC/MS联用分析常用的定量方法和色谱一样有三种,外加一种GC-MS特有方法:
(1) 归一化法。将样品中所有组分含量之和作为100,计算各个组分的相对百分含量,称为归一化法。计算公式:
![]()
式中 Wi——组分i含量;Ai——组分i的峰面积(或峰高);fi——组分i的质量校正因子。当fi为体积校正因子或摩尔校正因子时,结果分别为体积分数或摩尔分数。
(2) 外标标准曲线方法。用标准样品配制不同浓度的标样,在与待测样品完全相同的操作条件下,测得标样中各化合物的峰面积或峰高,得到响应因子:
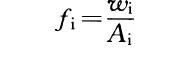
式中 Wi——待测化合物标样含量;Ai——待测化合物标样峰面积(或峰高)。
(3) 内标法内标标准曲线方法 。外标法存在一定缺点,限于每次的分析条件从样品处理到测试都会产生误差。为了克服不可避免的测量误差,选择适当的基准物质(内标化合物)加入标样和待测样品中进行测定,计算待测化合物和内标化合物响应值之比(称为相对响应因子),由相对响应因子和加入内标化合物的量进行定量,称为内标法。相对响应因子计算,可由:
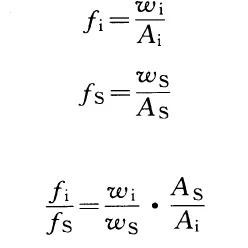
得到。式中 wi——待测化合物标样含量;Ai——待测化合物标样峰面积(或峰高);WS——内标化合物含量;As——内标化合物峰面积(或峰高)
(4)同位素稀释法。最理想的是采用待测化合物的同位素标记物作为内标,可以保证内标化合物的化学性质、色谱行为、质谱行为都与待测化合物一致,这样可以消除化合物之间的差别带来的误差。同位素标记物作内标只有GC/MS联用技术可以运用。计算公式:
![]()
式中,RF为校正因子,RF=含量/面积或含量/峰高;C为含量;S为峰面积或峰高。
同位素稀释法的优点为:一般可以校正基体影响(标样和样品的差别),提取效率稳定;进样误差小,保留时间随溶剂梯度变化影响不大;仪器漂移影响小;在质谱中同位素稀释剂与样品分析物响应稳定,定量结果从样品分析物与内标的响应比得出更准确。缺点为:样品制备很耗时;对移液和稀释等中的误差结果有缺陷。
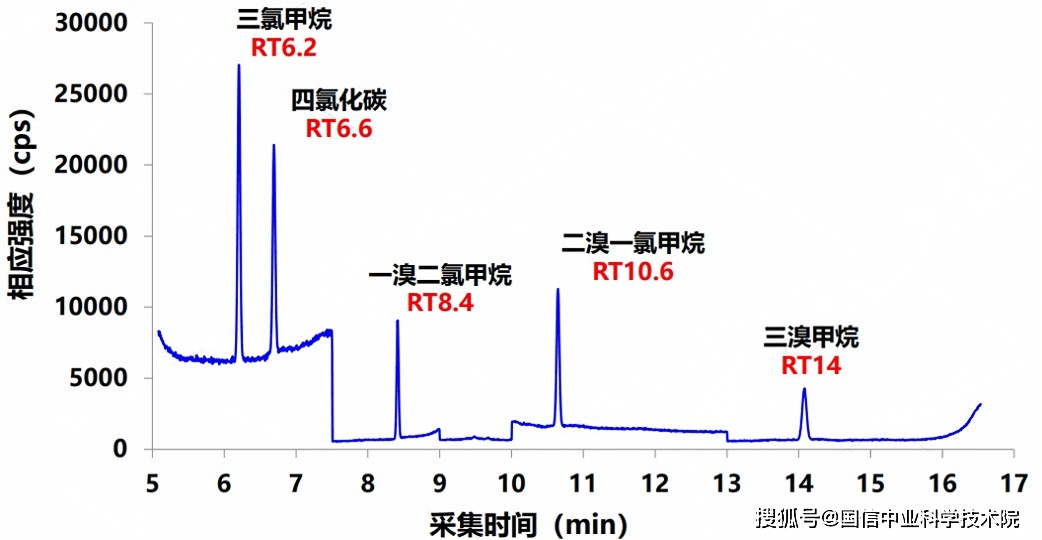
GC-MS应用:检测烃类 5种卤代烃
常见问题
1. 串联质谱如何定量?
答∶GCMS定量时,是以后面产生的碎片峰定量。因此,除填写对接单外,您需要在对接单中额外补充:
A、目标物质的标品-做外标;
B、目标物质的特征碎片-也称定量碎片;
C、目标物质的浓度范围-决定外标定量范围。
2. 色谱质谱类的定量测试,什么叫做标准品,什么叫做标点,什么叫做标准曲线(标曲)?
答:定量测试某种物质的浓度,一般需要纯度为99%该物质(称为标准品)作为溶质,配置一系列浓度梯度的溶液上机测试,配置的不同已知浓度的溶剂称为标点。不同浓度的溶液为X,峰面积为Y,拟合一元一次方程,称为标准曲线。未知浓度进样之后峰面积代入一元一次方程,计算浓度。
3. 样品含盐该怎么处理?
答∶根据样品类型,具体分为以下3种。
- 有机相液体样品:有机相进样一般不含盐,含盐建议前处理。如果您的样品含有盐,务必备注!!
- 水相液体样品:一般采用顶空进样、固相微萃取进样或者固相萃取成有机相再进样。其中,固相微萃取主要针对易挥发性的物质。如果采用固相萃取做为前处理,请将萃取步骤(包含但不限于溶剂、萃取柱的信息)一起填写。
PS:固相萃取小科普:优点是可以除盐、转化有机相、外加富集目标物质(后者在天然样品有机物定量过程中使用较广泛,因为一般天然样往往含量较低,低于检出限,但是富集之后可以在检出限内进行定量)。缺点是需要确认合适的萃取步骤并进行额外的前处理萃取工作,一般用C18小柱。GCMS测试目标产物比较低的时候(如自然水体内分泌物),可以先进行固相萃取,之后再进行GCMS测试。具体的萃取过程务必由客户提供,因为不同目标物质萃取步骤往往有所区别,根据目标物选择合适的萃取柱与萃取步骤,对检测结果的准确性有着至关重要的作用。
- 固体样品:如果样品可溶,可以选择将样品溶解在对应溶剂中,过滤上机;如果样品不可溶,比如说想要知道旧衣服的霉味是什么物质,可以采用固相微萃取进样(通过仪器升温系统促使有机物质挥发出来,再用SPME萃取纤维头进行吸附、富集,之后进样)。气体样品:一般是用铝箔采气袋送样,采用固相微萃取进样。气袋尽量装满,如用500mL的气袋装450mL气体是合理的,但是用1L的气袋装100mL的气体是不合理的。
4. GCMS柱流失是什么?
答∶所有的色谱柱都有柱流失的现象!!来源于固定相由于各种原因降解而产生的被洗脱物质,一般柱流失会随着温度的升高而加剧。GCMS柱流失成分主要是聚硅氧烷之类的物质,来自于色谱柱(柱流失成分并不是样品中的成分,所以整理结果的时候一般会直接筛选掉)。
5. 仪器的检出限是多少?
答∶正如毒理学中“抛开剂量谈毒性”的含义,GCMS检出限也并不是一个准确值。
检出限的定义为:某特定分析方法在给定的置信度内可从样品中检出待测物质的最小浓度或最小量。即,抛去其他细节因素,主要影响GCMS检出限的是测试方法以及对应测试物质。
举例:假如客户A的样品是液体样,需要检测其中四环素的含量。那么我们需要先按照测试方法设置好仪器,然后进标样,直到可以判定标品中存有浓度明显高于空白(大多是3倍噪声)的四环素出峰,这一标样浓度可以称为检出限。
如果在自身样品各种信息都没有提供的前提下,直接问仪器检出限是多少?就说明欠缺对检出限定义、GCMS基础原理的理解。
6. 质谱总离子流图TIC和自己在实验室做的GC图对不上?
答∶这也是对GCMS、GC原理理解不足,经常问出的问题。
GC可以后搭多种检测器,如热导检测器(TCD)、火焰离子化检测器(FID )、热离子化检测器( NPD)、电子捕获检测器(ECD)等。搭配质谱检测器的这类称为GCMS。两者本身的重点就有所偏重。前面几种检测器,在判定物质时,更依赖于标品定性,一般是默认一个峰为一个物质,因此对分峰要求较为严格;而GCMS由于其质谱检测器的灵敏性,重点在于碎片的区分。即使GCMS上显示是一个峰,也可以根据质谱数据判定前半个峰、后半个峰、甚至峰顶是否是同一个物质。
之前有客户需求,在自己实验室已经做过GC,有10个峰,但是不知道是什么物质,无法买对应标品去定性。希望我们在指南针GCMS仪器上帮他定出那10个峰具体是什么。
该需求暴露出客户缺乏专业知识:以自身实验室GC的方法强行套在GCMS仪器上。
GCMS仪器本身不是用于优化峰的仪器,强行优化实际是做一个方法学开发,费用非常高;即使强行优化,由于GCMS仪器不可能和客户实验室GC配置一模一样,强行进行也不能保证结果,风险较大。
7. 数据库对比能做吗?
定性时,GCMS一般使用NIST数据库进行对比查询,若需其他分析,客户自行处理或者沟通数据分析收费服务。如何进行筛选,不包括在测试服务中。定量时,不做NIST数据库筛选。
8. 气质电离方式有哪些?
答∶气质的电离方式一般有电子电离(EI)和化学电离(Cl)。其中,EI源最为常见,也是指南针默认电离方式。
9. GCMS匹配NIST数据库结果怎么看?
答:出峰用NIST数据库匹配。小峰不做筛选,因为强度比较弱,匹配不了。
定性一栏对应的是匹配度,匹配度越高,可信度就越高。如果某物质匹配度较低,通常认为该物质存在的可能性较低。数据库匹配(定性)只是提供了一个物质鉴定的参考或这个物质存在的可能性,作为一种定性的辅助手段,如果想准确确认建议走标品验证,并结合其他表征手段(比如核磁、红外等)来实现。
10. 什么是溶剂延迟,为什么设置溶剂延迟?
答:溶剂延迟是指测试时设置一段时间让溶剂完全流出色谱柱而不被质谱检测,即在谱图中会缺失前几分钟的数据。为什么要设置溶剂延迟呢?首先溶剂浓度一般比较高,设置溶剂延迟,可以有效保护质谱灯丝,延长质谱使用寿命;其次溶剂峰信号一般比较强,若不设置溶剂延迟,溶剂峰在谱图中的占比比较大,会使得其他成分(尤其是含量低的成分)信号峰比例很小,可能在整个谱图中显示不明显,不便于观察;再次最终结果中面积归一化法定量默认是针对所有峰的归一化定量,若不设置溶剂延迟,归一化定量也是包含溶剂峰等,若只关注非溶剂成分的归一化定量,需要扣除溶剂峰重新计算,增加后期数据分析的工作量。当然,以下几种情况是不建议设置溶剂延迟的:1)样品本身就是做溶剂分析;2)样品溶剂出峰靠后,目标成分出峰靠前;3)溶剂峰和目标成分分不开;4)需要得到样品中溶剂的归一化占比等。这几种情况若设置溶剂延迟可能会丢失一定的信息,并且设置溶剂延迟的信号后期是无法复原的,而不设置溶剂延迟,后期可以自己作图,把谱图原始数据中的溶剂峰数据删除掉,可操作空间会较大。
PS: 常规溶剂,比如甲醇、乙醇、乙腈、丙酮、正己烷、二氯甲烷、氯仿、乙酸乙酯等,这些溶剂一般出峰都是比较靠前的,若对溶剂峰不关注的话,一般都建议设置溶剂延迟。

免责声明:本资源来源于实验分析知识集中营,版权归原作者所有,本文仅作交流学习之用,不作任何商业用途,若有侵权,请联系删除,谢谢理解!
 感谢点赞
+1
感谢点赞
+1
