 预存有礼
预存有礼



第一性原理
 仪器型号:
仪器型号:
根据方案
 预约次数:
预约次数:5683次
 服务周期:
服务周期:15-20个工作日
 项目介绍
项目介绍第一性原理是指不使用经验参数,只通过基本物理常量(me、c、e、h、kB)来计算微观体系基本性质的方法。它包括基于密度泛函的从头算和基于Hartree-Fock自洽计算的从头算,前者以电子密度作为基本变量(Hohenberg-Kohn定理),通过求解Kohn-Sham方程,进行迭代自洽得到体系的基态电子密度,进而求得体系的基态性质;后者则通过自治求解Hartree-Fock方程,获得体系的波函数进而求得基态性质。
常用软件:VASP,MS,CP2K,QE等
计算内容包括但不限于
1.电子结构计算:电荷密度、电荷差分密度、态密度、能带、费米能级、功函数、ELF等;
2.几何结构计算:键长、键角、晶格常数、原子位置等;
3.材料性质计算:介电常数、弹性模量、磁矩、磁导率、热导率、界面热阻等;
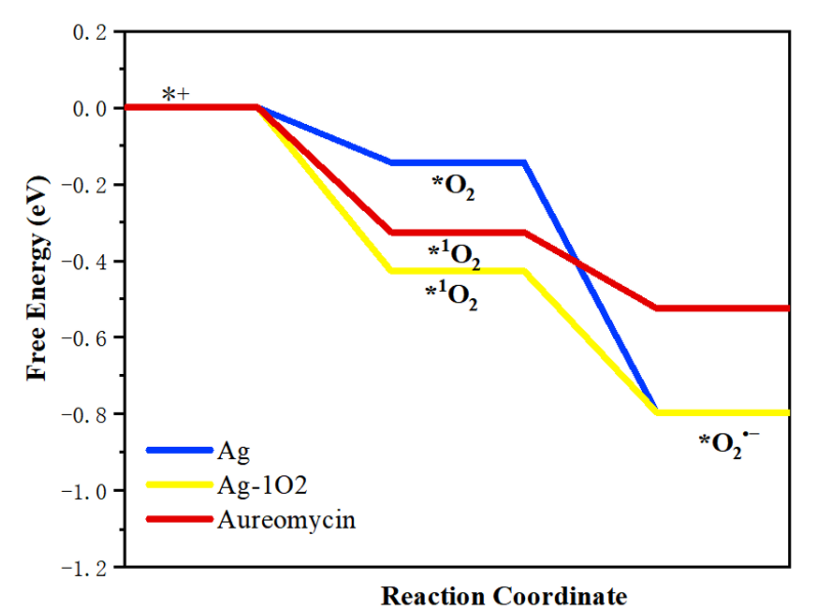
4.催化相关计算:如HER、OER/ORR、NRR、CO2RR等;
5.能量相关计算:如吉布斯自由能、过渡态、吸附能、剥离能、形成能等,
6.反应相关计算:如反应路径、反应机理研究、预测反应产物、能垒计算等;
7.其他计算:团簇、异质结、锂、钠、钾电池电极材料、锂-硫电池、碱金属离子电池、钙钛矿材料、骨架材料、高熵合金等DFT计算,
 结果展示
结果展示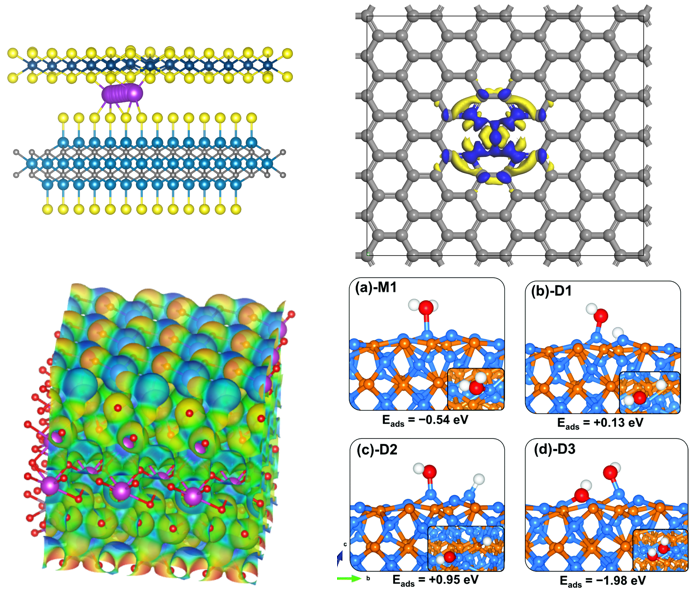
 常见问题
常见问题-
 为什么能带计算值一般低于实验值?
为什么能带计算值一般低于实验值?
这是PBE本身的问题,PBE是一种GGA泛函,有较强的自相互作用问题,导致电子偏向离域、带隙低估等一系列问题。一般计算得到的带隙比实验低30%~50%。这种低估并不是说方法就不可以用,我们还是可以把这个方法拿来做定性分析。 解决办法,方法有DFT+U、mBJ+U、杂化泛函

技术顾问

赵工 15757137609
扫码咨询技术顾问